
Sickle cell disease is the name given to a family of conditions. The most serious type is sickle cell anaemia (Hb SS). Symptoms include pain known as a ‘crisis’, severe anaemia, susceptibility to infections and damage to major organs.
Beta thalassaemia major is caused by a defect in the normal haemoglobin gene, which prevents the body from producing haemoglobin. The result is severe anaemia, and people need life-long treatment with regular blood transfusions for survival, and medicine to clear excess iron from the body.
Carriers for a haemoglobin disorder are healthy. They have inherited one unusual haemoglobin gene and one gene for normal haemoglobin A. Carriers are unaware of their status unless they have a specific blood test and they will never develop a haemoglobin disorder. But the gene is still there in the background, and they could pass it on to their own children.
Anyone can be a carrier of a haemoglobin disorder. It tends to be most common among people whose ancestors come from Africa, the Caribbean, the Mediterranean, some parts of India, Pakistan, south and south-east Asia and the Middle East. This is because carrying a gene for a haemoglobin disorder may give partial protection against malarial infection, so in places where malaria has been widespread, the genes have become more common. Carriers can still get malaria, and should always protect themselves when travelling.
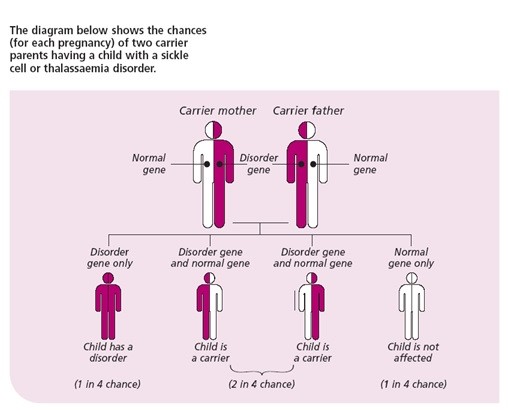
Where both parents are carriers, there is a 1 in 4 (25%) chance that their baby could inherit both unusual haemoglobin genes and have a condition that requires treatment.
Since 2008, all pregnant women in England (and the baby’s father where the woman is identified as a carrier), are offered screening for carrier status. All newborn babies are offered screening for sickle cell disease as part of the newborn blood spot (heel prick) test.
Sickle Cell &Thalassaemia Antenatal Screening Programme
The sickle cell and thalassaemia screening programme require women to be offered screening antenatally by 10+0 weeks of pregnancy. This is supported by the National Screening Committee and the National Institute of Health and Clinical Excellence, which state that women should access antenatal care by 10+0 weeks of pregnancy. In fact, early access to antenatal care is generally preferred by women.
Offering screening early in pregnancy
One of the reasons for encouraging early booking for antenatal care is to facilitate timely screening of the baby’s mother and father. Prenatal diagnosis (to identify if the baby has a haemoglobin disorder) should be offered by 12+6 weeks of pregnancy, if the woman/couple require it. This is really important, as it allows the couple sufficient time to make informed choices regarding the pregnancy.
There is a known link between when screening is offered and uptake of prenatal diagnosis (PND). An early offer of screening is linked to a greater uptake of PND. This is confirmed by evidence from research conducted within the UK population.
Early screening is challenging to achieve, given the variation in the practice of booking women for antenatal care throughout England. However, since establishment of targets to measure the quality of the screening programme, some hospitals have been working hard to improve early booking of women for antenatal care and screening.
Testing at other times
Blood tests for sickle cell and thalassaemia can be offered at any stage in life. It is helpful for people to know their carrier status before they plan a family. The test can be done by either a GP or at a specialist sickle cell and thalassaemia centre.
For further information
Public Health England
www.gov.uk/population-screening-programmes
www.gov.uk/government/organisations/public-health-england
Sickle Cell &Thalassaemia Screening Programme
www.gov.uk/topic/population-screening-programmes/sickle-cell-thalassaemia
Email: phe.screeninghelpdesk@nhs.net
Sickle Cell Society
www. sicklecellsociety.org/
UK Thalassaemia Society
www.ukts.org/
Patient information and resources
www.nhs.uk/conditions/thalassaemia/pages/symptoms.aspx
www.nhs.uk/Conditions/Sickle-cell-anaemia/Pages/Introduction.aspx